




%22 fill-rule=%22nonzero%22%3E%3Cpath d=%22M15.754367 31.4866226c-4.2011645.0-8.13699236-1.636243-11.14414171-4.5991696-6.14696705-6.146967-6.14696705-16.0970936.0-22.24406062C7.57315186 1.68046581 11.5532025.0442227846 15.754367.0442227846S23.8913594 1.68046581 26.8985087 4.64339238L21.6359973 9.90590374c-2.2995848-2.2995848-5.7489619-3.09559492-9.0656708-1.85735695C8.28071641 9.99434931 6.37913667 14.5492961 8.05960249 18.9715746c1.45935189 3.2282632 4.46650121 5.129843 7.73898731 5.129843 2.2111392.0 4.2896101-.8844557 5.8816303-2.4322532l5.2625114 5.2625114c-3.0071494 2.9187038-6.9872 4.5549468-11.1883645 4.5549468z%22 id=%22%E8%B7%AF%E5%BE%84%22 fill=%22%23001f60%22/%3E%3Cg id=%22%E7%BC%96%E7%BB%84%22 transform=%22translate(31.099673, 0.663342)%22%3E%3Cpolygon id=%22%E8%B7%AF%E5%BE%84%22 fill=%22%23001f60%22 points=%220 14.5050733 0 30.2041619 7.42942781 30.2041619 7.42942781 7.11986831%22/%3E%3Crect id=%22%E7%9F%A9%E5%BD%A2%22 fill=%22%2300bbb4%22 x=%220%22 y=%22-125688713e-22%22 width=%227.42942781%22 height=%227.11986831%22/%3E%3C/g%3E%3Cpath d=%22M99.6449893 19.0600201C98.1414146 13.6648404 92.525121 12.6919392 87.9701742 11.895929 87.5279463 11.8074835 87.0414957 11.7190379 86.5992679 11.6305923 83.5921185 11.0556961 83.3267818 10.878805 82.884554 10.6134683 82.0885438 10.0827949 82.0885438 9.06567084 82.4423261 8.49077464 83.0614451 7.47365059 84.6976881 6.89875439 86.6434907 6.89875439c4.0242733.0 5.2625113 1.7246886 5.4394025 2.74181264H99.7334349c0-2.43225315-1.4151291-4.95295187-3.75893670000001-6.76608603C94.2498096 1.54779746 91.2426602.0 86.5992679.0 81.4252021.0 78.4622755 1.94580252 76.914478 3.58204555 75.2340122 5.35095693 74.3495565 7.65054173 74.4380021 9.90590374 74.7033388 16.1855391 80.3638552 18.0428961 85.2725843 18.9273518 85.759035 19.0157974 86.2454856 19.1042429 86.7319362 19.1926885 87.9259514 19.4138024 89.4737489 19.6349163 90.7562096 20.0329214 91.5964425 20.2982581 92.4366754 21.0500455 92.0828932 22.2440606c-.2653368.8402329-1.6804659 1.9458026-4.2011646 2.1669165C85.2283615 24.632091 83.1056679 23.9687492 82.1327666 22.8631796 81.4694249 22.1556151 81.3809793 21.2711594 81.3809793 20.8289315H73.9515515C73.9515515 23.5707442 75.1013439 26.0914429 77.135592 27.9930226c2.3880303 2.2111393 5.8816303 3.4051544 9.6847898 3.4051544C87.3510552 31.398177 87.8817286 31.3539543 88.412402 31.3097315 92.8346805 30.9117264 96.3725033 29.142815 98.3183058 26.3567796 99.9545488 24.2783087 100.396777 21.6691644 99.6449893 19.0600201z%22 id=%22%E8%B7%AF%E5%BE%84%22 fill=%22%23001f60%22/%3E%3Cpath d=%22M71.6519667 30.8675036 63.7360882 20.2098125C67.9372528 19.0157974 70.9886249 15.2126379 70.9886249 10.6134683 70.9886249 5.12984301 66.5221237.663341768 61.0384984.663341768H44.8529592V30.8675036H52.282387V20.5635948h3.2282633l7.606319 10.3039088h8.5349974zM52.2381643 7.82743287h8.3581062c1.59202029999999.0 2.874481 1.28246075 2.874481 2.87448103.0 1.5920202-1.2824607 2.874481-2.874481 2.874481H52.2381643V7.82743287z%22 id=%22%E5%BD%A2%E7%8A%B6%22 fill=%22%23001f60%22/%3E%3C/g%3E%3C/g%3E%3C/svg%3E)







%22%3E%3Cg id=%22%E7%BC%96%E7%BB%84-34%22 transform=%22translate(1654.000000, 46.000000)%22%3E%3Cg id=%22%E7%9F%A9%E5%BD%A2%22 opacity=%22.01%22%3E%3Cuse fill=%22%23fff%22 xlink:href=%22%23path-1%22/%3E%3Cuse fill=%22%23f2f3f7%22 xlink:href=%22%23path-1%22/%3E%3C/g%3E%3Cg id=%22sousuo%22 transform=%22translate(6.000000, 6.000000)%22%3E%3Cmask id=%22mask-3%22 fill=%22%23fff%22%3E%3Cuse xlink:href=%22%23path-2%22/%3E%3C/mask%3E%3Cg id=%22Clip-2%22/%3E%3Cpath d=%22M11.6489827.0271704545c6.433526.0 11.6491213 5.1088636355 11.6491213 11.4108750455.0 2.9073068-1.110104 5.5606704-2.9376763 7.5756477C20.393341 19.0389886 20.424763 19.0662955 20.4543121 19.0953068l3.24437 3.1817727C24.099711 22.6703523 24.1004046 23.3084659 23.7004509 23.7025227L23.6986821 23.7042614C23.2966127 24.0985568 22.6454913 24.0985568 22.243422 23.7042614l-3.2445434-3.1817728C18.9553873 20.4797386 18.915815 20.433375 18.8807514 20.3838409L18.8809942 20.3841136c-1.986763 1.5431591-4.4995838 2.4646705-7.2320115 2.4646705C5.21545665 22.8487841.0 17.7400568.0 11.4380455.0 5.13603409 5.21545665.0271704545 11.6489827.0271704545zm0 2.0661136355c-5.26855495.0-9.53975727 4.18384091-9.53975727 9.34476141.0 5.1609204 4.27120232 9.344625 9.53975727 9.344625 5.2686936.0 9.5397225-4.1837046 9.5397225-9.344625.0-5.1609205-4.2710289-9.34476141-9.5397225-9.34476141z%22 id=%22Fill-1%22 fill=%22%23424242%22 mask=%22url(%23mask-3)%22/%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
%22%3E%3Cg id=%22%E7%BC%96%E7%BB%84-8%22 transform=%22translate(1165.000000, 90.000000)%22%3E%3Cg id=%22%E7%BC%96%E7%BB%84-5%22 transform=%22translate(30.000000, 35.000000)%22%3E%3Cg id=%22%E7%BC%96%E7%BB%84-6%22 transform=%22translate(413.000000, 3.000000)%22%3E%3Cg id=%22sousuo%22 transform=%22translate(12.000000, 12.000000)%22%3E%3Cmask id=%22mask-2%22 fill=%22%23fff%22%3E%3Cuse xlink:href=%22%23path-1%22/%3E%3C/mask%3E%3Cg id=%22Clip-2%22/%3E%3Cpath d=%22M14.6489827 3.02717045c6.433526.0 11.6491213 5.10886364 11.6491213 11.41087505.0 2.9073068-1.110104 5.5606704-2.9376763 7.5756477C23.393341 22.0389886 23.424763 22.0662955 23.4543121 22.0953068l3.24437 3.1817727C27.099711 25.6703523 27.1004046 26.3084659 26.7004509 26.7025227L26.6986821 26.7042614C26.2966127 27.0985568 25.6454913 27.0985568 25.243422 26.7042614l-3.2445434-3.1817728C21.9553873 23.4797386 21.915815 23.433375 21.8807514 23.3838409L21.8809942 23.3841136c-1.986763 1.5431591-4.4995838 2.4646705-7.2320115 2.4646705C8.21545665 25.8487841 3 20.7400568 3 14.4380455 3 8.13603409 8.21545665 3.02717045 14.6489827 3.02717045zm0 2.06611364c-5.26855495.0-9.53975727 4.18384091-9.53975727 9.34476141.0 5.1609204 4.27120232 9.344625 9.53975727 9.344625 5.2686936.0 9.5397225-4.1837046 9.5397225-9.344625.0-5.1609205-4.2710289-9.34476141-9.5397225-9.34476141z%22 id=%22Fill-1%22 stroke=%22%23f2fbff%22 fill=%22%23f5f5f5%22 mask=%22url(%23mask-2)%22/%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
%22%3E%3Cg id=%22%E7%BC%96%E7%BB%84-19%22 transform=%22translate(515.000000, 180.000000)%22%3E%3Crect id=%22%E7%9F%A9%E5%BD%A2%22 fill=%22%23fff%22 opacity=%22.2%22 x=%220%22 y=%220%22 width=%2280%22 height=%2280%22 rx=%2240%22/%3E%3Cg id=%22sousuo%22 opacity=%22.7%22 transform=%22translate(21.000000, 21.000000)%22%3E%3Cmask id=%22mask-2%22 fill=%22%23fff%22%3E%3Cuse xlink:href=%22%23path-1%22/%3E%3C/mask%3E%3Cg id=%22Clip-2%22/%3E%3Cpath d=%22M18.4442225.0430198864c10.1864162.0 18.4444422 8.0890340936 18.4444422 18.0672187136.0 4.6032358-1.7576647 8.8043949-4.6513208 11.9947756C32.2894566 30.1450653 32.3392081 30.1883011 32.3859942 30.2342358l5.1369191 5.0378068C38.1578757 35.8947244 38.158974 36.905071 37.5257139 37.5289943L37.5229133 37.5317472C36.8863035 38.1560483 35.8553613 38.1560483 35.2187514 37.5317472l-5.1371936-5.0378069C30.0126965 32.4262528 29.9500405 32.3528437 29.8945231 32.2744148L29.8949075 32.2748466C26.7491994 34.7181818 22.7705665 36.1772415 18.4442225 36.1772415 8.25780636 36.1772415.0 28.0884233.0 18.1102386.0 8.13205398 8.25780636.0430198864 18.4442225.0430198864zm0 3.2713465936c-8.3418786.0-15.10461556 6.62441477-15.10461556 14.79587212.0 8.1714574 6.76273696 14.7956563 15.10461556 14.7956563 8.3420983.0 15.1045607-6.6241989 15.1045607-14.7956563.0-8.17145735-6.7624624-14.79587212-15.1045607-14.79587212z%22 id=%22Fill-1%22 fill=%22%23000%22 mask=%22url(%23mask-2)%22/%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
%22%3E%3Cg id=%22%E7%BC%96%E7%BB%84-11%22 transform=%22translate(625.000000, 180.000000)%22%3E%3Cg id=%22%E7%9F%A9%E5%BD%A2%22 opacity=%22.1%22%3E%3Cuse fill=%22%23fff%22 xlink:href=%22%23path-1%22/%3E%3Cuse fill=%22%23000%22 xlink:href=%22%23path-1%22/%3E%3C/g%3E%3Cg id=%22%E5%BD%A2%E7%8A%B6%E7%BB%93%E5%90%88-2%22 opacity=%22.9%22 transform=%22translate(23.000000, 23.000000)%22 fill=%22%23fff%22%3E%3Cpath d=%22M3.41421356.585786438 16.849 14.02 30.2842712.585786438C31.024212-.154154326 32.1996962-.193098576 32.9854836.468953686L33.1126984.585786438C33.893747 1.36683502 33.893747 2.63316498 33.1126984 3.41421356L19.677 16.849 33.1126984 30.2842712C33.893747 31.0653198 33.893747 32.3316498 33.1126984 33.1126984s-2.0473786.781048599999998-2.8284272.0L16.849 19.677 3.41421356 33.1126984C2.6742728 33.8526391 1.49878864 33.8915834.713001205 33.2295311L.585786438 33.1126984c-.781048584-.781048599999998-.781048584-2.0473786.0-2.8284272L14.02 16.849.585786438 3.41421356c-.781048584-.78104858-.781048584-2.04737854.0-2.828427122.781048582-.781048584 2.047378542-.781048584 2.828427122.0z%22 id=%22%E5%BD%A2%E7%8A%B6%E7%BB%93%E5%90%88%22/%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/svg%3E)

 為加強對藥械組合產品注冊工作的監督和指導,進一步鼓勵具有臨床價值的藥械組合產品上市,構建適合我國國情的藥械組合產品的管理模式,國家藥品監督管理局將藥械組合產品技術評價作為監管科學研究項目,組織制定了《以醫療器械作用為主的藥械組合產品注冊審查指導原則》《以醫療器械作用為主的藥械組合產品中藥物定性定量及體外釋放研究注冊審查指導原則》,現予發布。
為加強對藥械組合產品注冊工作的監督和指導,進一步鼓勵具有臨床價值的藥械組合產品上市,構建適合我國國情的藥械組合產品的管理模式,國家藥品監督管理局將藥械組合產品技術評價作為監管科學研究項目,組織制定了《以醫療器械作用為主的藥械組合產品注冊審查指導原則》《以醫療器械作用為主的藥械組合產品中藥物定性定量及體外釋放研究注冊審查指導原則》,現予發布。特此通告。
附件:1.以醫療器械作用為主的藥械組合產品注冊審查指導原則
2.以醫療器械作用為主的藥械組合產品中藥物定性定量及體外釋放研究注冊審查指導原則
國家藥監局
2022年1月11日
【附件1】
以醫療器械作用為主的藥械組合產品注冊審查指導原則
以醫療器械作用為主的藥械組合產品(本指導原則簡稱為藥械組合醫療器械),預期可能使產品在疾病的預防、治療等過程中更加安全和有效,但也可能會引發新的技術關注點,因此為了進一步指導申請人對藥械組合醫療器械注冊申報的準備,鼓勵該類產品的創新發展,制訂本指導原則。
本指導原則系對藥械組合醫療器械注冊審查的一般指導文件。由于該類產品品種多樣,本指導原則主要對藥械組合醫療器械中藥物相關要求提供了指導,申請人可依據具體產品的特性對資料進行充實和細化。本指導原則雖然為該類產品的注冊申報提供了初步指導和建議,但不會限制醫療器械相關管理部門及該類產品的技術審評、行政審批,以及申請人對注冊申報資料的準備。
本指導原則是在現行法規以及當前認知水平下制訂的,隨著法規的不斷完善,以及藥械組合醫療器械技術的發展和提高,本指導原則相關內容也將進行適時調整。
一、適用范圍
本指導原則涉及的藥械組合產品系指由藥品與醫療器械共同組成,并作為一個單一實體生產的醫療產品,其中以醫療器械作用為主的藥械組合產品按照醫療器械管理,簡稱藥械組合醫療器械。
本指導原則適用于藥械組合醫療器械注冊申報資料的準備和注冊審查。本指導原則僅對產品中藥物部分要求提出了建議。本指導原則不涉及藥械組合醫療器械的界定程序及要求。
二、基本要求
藥械組合醫療器械本質上為醫療器械產品,應按照《醫療器械注冊申報資料要求及說明》準備相關注冊申報資料,并在注冊申請表中注明“藥械組合產品”。
申請人應將藥械組合醫療器械作為單一實體評估產品的安全性和有效性,需特別關注藥物和/或醫療器械與藥物相互作用等引入的潛在風險。申請人可結合已有的藥物相關信息進行評估,但藥械組合醫療器械中藥物的安全性和有效性可能不同于單獨使用的藥物。通常藥械組合醫療器械重點關注藥物局部應用的安全性和有效性,但當組合應用后藥物血藥濃度大于常規藥物使用的血藥濃度時,也需對系統毒性進行評價。
必要時,建議申請人同時參考適用的藥品相關指導原則。
三、特殊要求
(一)電子申報資料項目提交要求
申請人應按照醫療器械注冊電子申報系統(eRPS)格式要求準備相關申報資料,并在CH3.8(其他資料)提交藥物相關產品描述、藥物和/或醫療器械與藥物相互作用、藥物含量/劑量選擇資料。同時在CH3.8項提交相關研究總結,若具體研究資料在其他eRPS項目提交,需明確相應的eRPS項目編號及文件名稱。如適用,建議其他項目中藥物相關資料以附件形式提交,資料名稱宜參照以下命名規則:
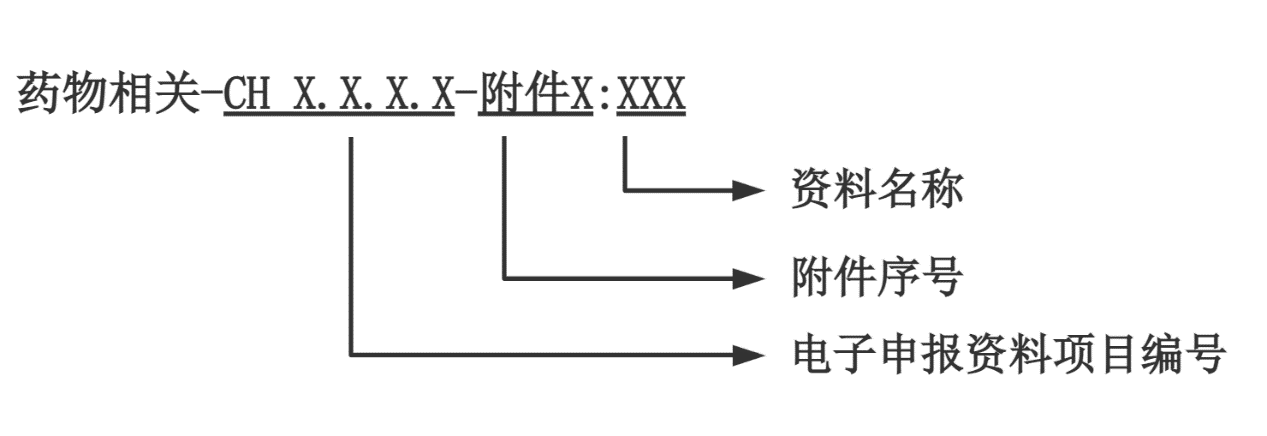
圖1 文件命名規則
舉例:藥物相關-CH3.7.1 產品穩定性-附件1:實時老化研究報告
(二)產品描述
申請人需詳細描述藥械組合醫療器械產品中藥物名稱、預期使用目的、首要作用方式、來源及相關許可文件(如有)。
1.藥物基本信息
申請人需明確產品中藥物的名稱。適用時,提供相關信息如化學結構式、分子量、性狀、純度、含量、劑量(如μg/mm2)、載體類型、與載體的配比、藥物與器械結合方式等;提供理化性質參數如色澤、pH值、解離常數(pKa)、粒度、晶型、比旋度、光學異構體、熔點、水分、溶解度等;提供穩定性信息如對光、濕、熱的穩定性,固、液態下的穩定性和相互作用的穩定性;提供藥物已有的吸收、分布、代謝、消除等信息。如已上市藥物發生相關不良事件或召回事件,建議提供相關信息。
如藥物已獲得我國或生產國(地區)批準上市銷售的,應提供上市銷售證明性文件。如已有我國或生產國(地區)批準上市銷售的藥物,建議優先采用,未采用宜說明理由。如原料藥已在某種或某些藥物上市時通過關聯審評或進行了主文檔備案,建議提交關聯/備案信息說明。
2.預期使用原因
申請人需提供采用藥械組合方式的合理性和必要性分析,如臨床治療需求,分析論述添加藥物成分的受益和新增風險,提供相關支持性資料。如已有可參考的同類產品或前代產品,宜提供同類產品或前代產品信息,說明與其差異并評估影響。如無明確證據表明適合開發為藥械組合醫療器械,不宜采用藥械組合方式。
3.作用方式
作用方式為產品實現預期治療效果或作用的手段,藥械組合醫療器械以醫療器械作用方式為主,藥物在醫療器械主要作用基礎上發揮作用。申請人需闡明藥物在醫療器械中發揮作用的原理,實現預期適用范圍的機理和作用的持續時間,并提供支持性資料。
如作為藥械組合醫療器械,產生新的作用機理、新的適應證、新的目標人群、新的使用方法等,申請人需予以說明。
(三)醫療器械與藥物的相互作用
申請人通過化學、物理或其他方式等制造藥械組合醫療器械產品時,宜在設計開發全過程中考慮醫療器械和藥物間的潛在相互作用,如器械/藥物性能間的相互增強或削弱。例如藥物洗脫支架的藥物涂層對支架的置入、生物相容性會產生影響;含藥骨水泥的混合聚合過程可能對藥物有效性產生影響。一些藥械組合醫療器械可能會出現增效作用;一些藥物由于器械能量發出而降低穩定性或活性;一些藥物放射性導致器械材料退化,因此對于藥械組合醫療器械而言,單獨的器械部分或藥物部分評估可能并不能充分評估產品的安全性和有效性,需進一步考慮兩者間的相互潛在作用的影響并提供研究資料。
(四)藥物含量/劑量選擇
申請人需提供藥械組合醫療器械中藥物的含量/劑量選擇/確定依據,如需控制藥物釋放的,還需提供配方信息如比例以及配方篩選依據。當參考前代產品或同類產品選擇含量/劑量,需評估產品設計差異的影響,如釋放速率不同。
(五)化學和物理性能
申請人需對藥物潛在引入的化學、物理性能風險進行研究/評估,如單體或溶劑殘留、添加劑/助劑/催化劑/交聯劑等殘留、藥物含量、劑量/生物學活性、純度、雜質、藥物釋放特性、涂層完整性、涂層耐久性、微粒等。
藥物若為申請人制備,如合成藥物、生物制品、中藥,申請人可參考藥物相關藥學研究指導原則提供資料,若申請人認為不必提交藥學研究指導原則建議的某項或某些資料,應標明不適用,并提出充分依據。
(六)生物學特性
對于藥械組合醫療器械,參考GB/T16886系列標準對終產品開展生物學評價,必要時開展生物學試驗,宜考慮藥物成分與試驗系統的兼容性。藥物可能會影響生物學試驗的結果,申請人可結合藥物的作用方式、臨床風險/受益論證藥物引入的生物學風險是否可接受。
對于某些藥械組合醫療器械,由于其中的藥物接觸時間可能不同于器械,所以僅采用一種接觸時間方式評估生物學風險可能是不合理的,如結合藥物的持續作用時間、藥物代謝/半衰期等,必要時補充評估藥物引入的長期/持久接觸生物學風險。
一般藥械組合醫療器械中藥物發揮局部作用,當產品含有某種已經獲得批準用于其他用途的藥物時,申請人可結合藥物安全性資料評價產品的安全性,但需考慮新的組合方式是否會使已經確定的或已經了解的安全性、有效性發生改變。如產品使局部或系統的藥物暴露大于已經批準的藥物劑量范圍,則可能還需要進一步補充安全性研究,如顱內應用器械可能導致的局部神經毒性等。
對于某些藥械組合醫療器械,可能需開展其他必要的試驗以評估藥物自身特殊的生物學風險(毒理學風險),如局部毒性試驗、光敏毒性試驗、依賴性試驗、致突變試驗或其他毒理學試驗。不需額外開展試驗的,可說明理由。
(七)動物試驗研究
申請人可依據《醫療器械動物試驗研究注冊審查指導原則第一部分:決策原則》決策是否需開展動物試驗研究。開展動物試驗研究時宜符合《醫療器械動物試驗研究注冊審查指導原則第二部分:試驗設計、實施質量保證》中相關建議。
醫療器械動物試驗是根據試驗目的,選用符合試驗要求的動物,在預先設計研究方案規定下,進行產品可行性和/或安全性和/或有效性研究,觀察、記錄動物的反應過程及結果,以確認醫療器械對生命活動的作用與影響。通常不需單獨開展試驗研究藥物自身的藥效學、藥代動力學,一般在醫療器械動物試驗中進行合并評價,并可設定藥物相關的觀察指標,宜可觀察到藥械組合醫療器械中藥物的有效性,但并不是藥效學研究。如有相關臨床文獻等資料,可一并提交論證。
適當時,宜對藥械組合醫療器械中藥物的體內外藥物釋放動力學和體內的藥代動力學開展研究。雖然藥械組合醫療器械的血藥濃度通常遠遠低于單獨作為藥物使用后的濃度,但局部組織濃度可能會遠遠高于藥物的血藥濃度,宜特別關注局部組織濃度變化情況。體內的藥代動力學研究需考慮多個產品聯合應用的情形如支架重疊使用,宜評估最高藥物暴露劑量水平下的藥代動力學。
藥械組合醫療器械中所含藥物具有耐藥性風險時,宜考慮耐藥性對產品有效性及公眾健康的影響。
(八)穩定性研究
適當時,申請人可參考《無源植入性醫療器械貨架有效期注冊申報資料指導原則》提供產品的穩定性研究資料,對于組合產品中藥物相關的穩定性,建議參考藥品相關指導原則進行穩定性研究。
(九)產品技術要求
申請人需在技術要求中明確藥物的名稱、含量/劑量、分子量、化學結構式,宜根據藥物的作用方式制定藥物適用的指標要求,如藥物定性、藥物含量(生物活性)、體外藥物釋放、藥物與器械結合牢固度、溶劑殘留等。對于不能進行客觀判定的或不屬于成品的功能性、安全性指標,不需在產品技術要求中制定。
(十)生產制造信息
申請人宜根據藥物及藥物載體的理化特性和藥物與載體的相互作用,選擇合適的藥物及藥物載體,并制定合理的生產工藝,規定有效的質量控制措施及控制指標。申請人需在生產制造信息中明確生產工藝流程及關鍵工藝和特殊工藝控制點,宜提供藥物與醫療器械結合工藝的研究資料,提供加工工藝如滅菌對藥物性能影響的研究資料。由于需對生物制品的生產過程和中間產品進行特殊控制,當藥械組合醫療器械中所含藥物為生物制品時,宜參照生物制品相關導則開展研究。
(十一)其他
如產品中所含藥物為麻醉、精神藥物或放射性藥物,應符合我國相關規定。
產品說明書中應明確所含藥物,并增加藥物過敏等風險提示。
四、臨床評價要求
藥械組合醫療器械的臨床評價應遵循組合產品研究和開發的基本規律,通過科學的過程來評估產品臨床療效和潛在風險,最終確定產品在預期用途下的安全性和有效性,并為產品使用說明書的撰寫提供依據。
器械上添加藥物成分,目的常為改善產品功能或減少與產品使用相關的不良事件等,建議提交支持資料論證其臨床風險/受益。
在進行臨床評價前,建議申請人明確組合產品的臨床作用機理、預期用途、可能帶來的風險、可能出現的不良事件等,并在臨床評價時予以充分考慮,如:組合產品的器械部分或藥物部分單獨使用時的適用范圍是否與組合產品的擬申報適用范圍一致、類似或存在較大差異;組合產品是否擴大或超出了其組成部分原單獨使用的適用范圍,或宣稱了更多的臨床受益;對于使用組合產品的患者,組合產品所含藥物部分的給藥途徑、釋放或局部/系統藥物暴露范圍等與藥物單獨使用時相比是否發生改變,以及其改變可能帶來的新增風險。
申請人可參考《醫療器械臨床試驗質量管理規范》《醫療器械臨床評價技術指導原則》《醫療器械等同性論證技術指導原則》《決策是否開展醫療器械臨床試驗技術指導原則》等相關要求開展組合產品的臨床評價。建議申請人結合組合產品的風險和受益,選擇合理的臨床評價路徑,論證其臨床應用的安全有效性。
組合產品的臨床試驗設計常涉及的主要問題包括樣本量、統計方法、臨床終點、適用范圍/功效宣稱、以及臨床研究數量(若包含多個試驗)等。在確定樣本量、統計方法、觀察指標、臨床終點、評估藥物-器械的相互作用時,建議結合組合產品的性能特點,論證組合產品臨床研究方案設計的科學性和充分性。
五、參考文獻
[1]國家市場監督管理總局.藥品注冊管理辦法 [EB],2020.
[2]U.S. Food and Drug Administration. Guidance for Industry and FDA Staff:Early Development Considerations for Innovative Combination Products [EB],2006.
[3]U.S. Food and Drug Administration. Technical Considerations for Pen, Jet, and Related Injectors Intended for Use with Drugs and Biological Products [EB],2013.
[4]國家藥品監督管理局.化學藥品注冊分類及申報資料要求[EB],2020.
[5]國家藥品監督管理局.生物制品注冊分類及申報資料要求[EB],2020.
[6]原國家食品藥品監督管理總局.化學藥物(原料藥和制劑)穩定性研究技術指導原則(修訂)[EB],2015.
六、起草單位
國家藥品監督管理局醫療器械技術審評中心
【附件2】
以醫療器械作用為主的藥械組合產品中藥物定性、定量及體外釋放研究注冊審查指導原則
目前,以醫療器械作用為主的藥械組合產品(本指導原則簡稱為藥械組合醫療器械)中,藥物與醫療器械組合形式主要是將藥物與醫療器械進行物理結合(如混合、涂覆等)或化學鍵合等。常見的藥械組合醫療器械有帶藥物涂層的支架、帶抗菌涂層的導管、含藥避孕套、含藥節育環等。
本指導原則旨在幫助和指導申請人開展藥械組合醫療器械產品注冊申報資料中的藥物定性、定量及體外釋放研究,以滿足技術審評的基本要求,同時有助于審評機構對該類產品進行科學規范的審評,提高審評工作的質量和效率。
本指導原則雖然為該類產品提供了初步指導和建議,但不會限制醫療器械相關管理部門及該類產品的技術審評、行政審批,以及申請人對注冊申報資料的準備。由于該類產品品種多樣,申請人可依據具體產品的特性對資料進行充實和細化。
本指導原則是在現行法規以及當前認知水平下制訂的,隨著法規的不斷完善,以及藥械組合醫療器械產品技術的發展和提高,本指導原則相關內容也將進行適時地調整。
一、適用范圍
本指導原則涉及的藥械組合產品系指由藥品與醫療器械共同組成,并作為一個單一實體生產的醫療產品,其中以醫療器械作用為主的藥械組合產品按照醫療器械有關要求申報注冊,簡稱藥械組合醫療器械。
本指導原則適用于藥械組合醫療器械的藥物定性、定量及體外釋放研究,可為此類產品的研發、注冊申報、技術審評等環節提供參考。
本指導原則不適用于以藥品作用為主的藥械組合產品。
二、研究內容
保證藥械組合醫療器械的安全、有效、質量可控是研發和評價應遵循的基本原則。藥械組合醫療器械中,所含藥物起輔助作用,需整體評估組合產品的新增風險和風險受益比,充分考慮藥物部分和器械部分的相互作用帶來的影響,考慮藥物部分較其單獨使用時給藥途徑、劑量、預期作用等可能發生變化及帶來的影響,因此建議基于藥物和醫療器械的組合形式及臨床預期用途,對該類產品進行藥物相關性能的研究,如藥物的定性、定量及體外釋放等。
一般情況下,藥械組合醫療器械產品需進行藥物定性、定量研究。對于通過將藥物釋放到預期部位(如采用緩釋、控釋或其他釋放方式)而發揮效用的產品,如帶藥物涂層的支架、帶藥球囊擴張導管、含銀敷料等,需進行藥物體外釋放研究。對于不通過將藥物釋放到預期部位而發揮效用的產品,如通過共價鍵結合添加肝素的人工血管等,則無需進行藥物體外釋放研究,但可通過體外釋放研究評估產品的穩定性、涂層牢固度等。
添加有生物活性物質的產品,如含生物活性物質的骨科器械和含肝素涂層的器械,需根據其作用機理開展相關研究,對主要成分進行鑒定,可通過含量、活性、效價等形式的研究進行定性、定量。
申請人可根據產品的組合形式、預期用途、藥物作用方式等參照此原則,選擇合適的研究內容。
本指導原則是對藥械組合醫療器械藥物定性、定量及體外釋放研究的一般要求,對于具體產品,若有專門的指導原則或標準,建議參考相應的指導原則或標準的具體要求。申請人應依據組合產品的具體特性和研究目的對注冊申報資料的內容進行充實和細化,并對在研究過程中評價技術的設計、實施及結果應用的科學性和合理性進行充分的闡述。
三、藥物定性、定量、體外釋放及方法學驗證
(一)藥物定性、定量
藥物定性的目的在于確定被分析物是目標物,而非其它物質。用于鑒別的分析方法應具有較強的專屬性。藥物定量的目的在于準確測定藥械組合醫療器械產品中所載藥物的量。常用的分析方法包括理化方法及生物學方法等。
藥物定性、定量分析方法應優先選用標準方法,如國際標準、國家標準等,在使用上述方法之前,可參照中國藥典四部中9099《分析方法確認指導原則》、9100《分析方法轉移指導原則》和9101《分析方法驗證指導原則》等指導原則中規定的方法進行方法確認或轉移,以確保方法的適用性。如果無適用方法,可參照由知名技術組織或有關科技文獻或期刊中公布的方法,開發適宜的新方法并進行全面的方法學驗證。在某些情況下,如原材料的合成工藝改變、分析方法中某些參數發生改變如色譜柱型號、流動相、柱流速等,應考慮是否需要對分析方法再次進行全面的或部分的驗證,以確保分析方法可行。
對于可以直接在器械上或采用產品原液進行藥物定性和定量進行試驗的產品,可直接進行產品的藥物定性、定量研究;需將藥物從醫療器械中浸提(洗脫或分離)出來或適當稀釋后進行試驗的產品,提供浸提液/稀釋液制備及分析方法確定的依據。
建議申請人根據藥物性質、組合形式、器械材質等產品特性,選擇合適的處理方法并論述其適用性。對于需浸提后進行分析的產品,浸提液制備及分析方法可參照以下內容開展研究。
1.浸提液的制備
選擇終產品進行試驗,浸提樣品宜盡可能選取所有含藥部位,如截取含藥組件的部分進行浸提,需論述其合理性。考察因素通常包括:
(1)浸提溶劑:需考慮藥物的溶解性、穩定性等,尤其是具有生物活性的物質在浸提液制備過程中應避免其活性的改變。
(2)浸提比例:宜使溶液中藥物濃度保持在適宜的范圍內。
(3)浸提程度:適用的制備方法應確保所含藥物洗脫完全,以達到極限浸提為原則。
(4)浸提方式:可采取靜置、振蕩、超聲、回流等方式。必要時,可根據不同分析儀器及方法的要求,對浸提液處理(如離心、過濾等)后進行測定。
除此之外,基于分析手段的多樣性,也可先對藥物進行衍生、酸解、酶解等處理后,進行測定。
2.分析方法
根據產品中所含藥物的種類及分子結構、理化性質、生物活性等,選擇適宜的定性、定量方法。藥物定性一般與定量同時進行,常見的分析方法包括高效液相色譜法(HPLC)、紫外-可見分光光度法(UV-Vis)等。如含有紫杉醇、雷帕霉素、鹽酸利多卡因等藥物的醫療器械,可采用上述兩種方法進行分析。
采用高效液相色譜法時,可以通過保留時間(tR)和/或光譜相似度定性。用于定量時,可通過測定峰面積,使用內標法或外標法等進行計算。
采用紫外-可見分光光度法時,可以通過特定波長范圍內光譜圖、最大吸收波長(λmax)或兩個特定波長處的吸光度比值而定性。用于定量時,可在特定波長處測定吸光度,使用對照品比較法或吸收系數法等進行計算。
除此之外,其他的分析方法也可用于藥物的定性、定量分析,如藥物官能團的特異性反應、原子吸收分光光度法(AAS)、電感耦合等離子體發射光譜(ICP)、氣相色譜法(GC)、質譜法(MS)等,如含銀的醫療器械,可采用原子吸收分光光度法、電感耦合等離子體發射光譜法或電感耦合等離子體發射光譜-質譜法進行分析。
(二)藥物體外釋放
藥物體外釋放是在適宜的條件下,對藥械組合醫療器械進行的藥物體外釋放速率及釋放量的試驗。其目的是考察藥械組合醫療器械中藥物在體外的釋放情況及其規律,對生產工藝和產品質量進行控制,同時也可為動物試驗和臨床評價提供參考。
合理的藥物體外釋放行為是藥械組合醫療器械質量控制的重要指標,對于保證產品臨床使用的安全性和有效性具有重要意義。體外釋放試驗宜論證與體內釋放試驗的關聯性,考慮產品臨床使用情況,也可根據試驗需求,進行加速釋放試驗。
一般情況下,藥物體外釋放試驗通過測定藥械組合醫療器械在各時間點藥物的釋放率,來表征藥物的體外釋放行為。釋放率可通過釋放到介質中藥物的含量和/或釋放試驗后產品上剩余藥物含量計算而得。
在藥物體外釋放試驗中,應至少考慮以下因素:
1.試驗樣品
選擇終產品進行試驗,宜盡可能選取所有含藥部位,如截取含藥組件的部分進行浸提應論述其合理性。可適當增加測試樣品數量以降低樣品差異性引起的數據偏差。
2.釋放溫度
根據產品的預期使用部位,參考各國藥典中相應內容,選擇合適的溫度。如無特殊要求,通常選擇 37 ℃ ±1 ℃模擬人體正常體溫,32 ℃±1 ℃模擬表皮溫度。
3.釋放介質
根據藥械組合醫療器械預期使用部位的生理環境以及藥物理化性質等因素確定釋放介質,如生理鹽水、磷酸鹽緩沖液,或添加表面活性劑、有機溶劑、防腐劑、含相關生物酶的釋放介質、血清等。
4.時間點設置
時間點的設置需充分考慮產品中藥物在臨床的釋放情況。對于需要在體內快速釋放的藥物(如帶藥球囊擴張導管等)可設置一個時間點考察體外釋放情況。對于需要緩釋的器械(如帶藥物涂層的支架等),建議至少涵蓋藥物釋放的初始階段、中間階段和最后階段。初始階段為藥物釋放的活躍期,用于考察是否存在突釋效應;中間階段用于確定釋藥特性;最后階段表現藥物釋放的穩定期(又稱平臺期),用于考察藥物釋放是否完全,如冠狀動脈藥物洗脫支架一般要求至少釋放80 %標稱藥物含量時達到平臺期。如果體外釋放預期為非全部釋放,釋放時間宜考慮能覆蓋藥物洗脫量達到穩定的時間點。
藥物體外釋放試驗中,藥物的測定方法一般與定量方法一致,也可采用其他經驗證的方法。如果藥物為避光藥物,應關注藥物的避光要求。
(三)方法學驗證
方法學驗證的目的是證明建立的方法適合于相應檢測的要求,根據研究目的,可能需要的驗證指標有專屬性、準確度、精密度、檢測限、定量限、線性、范圍、耐用性等。對于含有生物活性物質的生物學測定方法驗證需參照相關指導原則開展方法學驗證。
需要說明的是,在方法開發和驗證過程中,需要考慮基質效應的影響。基質效應指的是在對分析物的濃度或質量測定過程中,來自樣品中一種或幾種其他化合物的綜合影響。需驗證在采用開發的方法對藥物進行測定時,不會因其他成分的存在而對擬研究藥物造成干擾。
1. 專屬性
專屬性系指在其他成分(如雜質、降解產物、基質等)可能存在下,采用的分析方法能正確鑒定、檢測出被測物的能力。宜采用適宜的方法對專屬性進行驗證,并排除其他成分的影響,如色譜方法中,應附典型圖譜,標明被測物的位置,且分離度符合相應要求。
藥物定性、定量、體外釋放研究的測定方法,均應考察其專屬性。如專屬性不強,應采用一種或多種不同原理的方法予以補充。
2. 準確度
準確度系指用所建立方法測定的結果與真實值或參比值接近的程度,通常用回收率來評價。準確度應在規定的線性范圍內試驗。
用于回收率試驗的樣品宜采用不含待測成分的空白樣品,可以采用加標回收率試驗。根據分析目的和樣品的濃度范圍,在以下兩種方法中任選其一:(1)在規定范圍內,取同一濃度(相當于100 %濃度水平)的供試品,用至少6份樣品的測定結果進行評價;(2)設計3種不同的濃度,覆蓋線性范圍的高、中、低濃度并考慮樣品的濃度范圍,每種濃度分別制備至少3份供試品溶液進行測定,計算回收率。
不同分析水平的可接受回收率也不同。樣品中待測成分含量和回收率限度關系可參考表1。在基質復雜、組分含量較低及多成分等分析中,回收率限度可適當放寬。
表1 藥械組合醫療器械中藥物含量和回收率限度
待測成分含量 | 回收率限度(%) |
1000 μg/mL | 90~108 |
100 μg/ mL | 85~110 |
10 μg/ mL | 80~115 |
1 μg/ mL | 75~120 |
0.01 μg/ mL | 70~125 |
3. 精密度
精密度系指在規定的測定條件下,同一份均勻供試液,經多次取樣測定所得結果之間的接近程度。精密度一般用偏差、標準偏差或相對標準偏差表示。
藥物定量測定應考察方法的精密度。根據分析目的和樣品的濃度范圍,在以下兩種方法中任選其一:
(1)在規定范圍內,取同一濃度(相當于100 %濃度水平)的供試品,用至少6份樣品的測定結果進行評價。
(2)設計3種不同的濃度,覆蓋線性范圍的高、中、低濃度并考慮樣品的濃度范圍,每種濃度分別制備至少3份供試品溶液進行測定。精密度的數據結果應報告標準偏差、相對標準偏差或置信區間。樣品中待測成分含量和精密度可接受范圍可參考表2。在基質復雜、組分含量低于0.01 %及多成分等分析中,精密度限度可適當放寬。
表2 藥械組合醫療器械中藥物含量與精密度可接受范圍關系
待測成分含量 | 重復性(RSD,%) |
1000 μg/mL | 3 |
100 μg/mL | 4 |
10 μg/mL | 6 |
1 μg/mL | 8 |
0.01 μg/mL | 15 |
4. 檢測限
檢測限系指樣品中被測物能被檢測出的最低量。檢測限僅作為定性鑒別的依據,沒有定量意義。常用的方法如下:
(1)直觀法
用已知濃度的被測物,試驗出能被可靠地檢測出的最低濃度或量。
(2)信噪比法
適用于能顯示基線噪音的分析方法,即把已知低濃度試樣測出的信號與空白樣品測出的信號進行比較,確定出能被可靠地檢測出的被測物質最低濃度或量。一般以信噪比為3:1或2:1時相應濃度確定為檢測限。
(3)基于響應值標準偏差和標準曲線斜率法
按照LOD=3.3δ/S公式計算,式中LOD為檢測限、δ為響應值的偏差、S為標準曲線的斜率。δ可以通過下列方法測得: = 1 \* GB3 ①測定空白值的標準偏差; = 2 \* GB3 ②采用標準曲線的剩余標準偏差或是截距的標準偏差。以上計算方法獲得的檢測限數據須用含量相近的樣品進行驗證。附測定圖譜,說明試驗過程和檢測限結果。
5. 定量限
定量限系指試樣中被測物能被定量測定的最低量,其測定結果應符合準確度和精密度的要求。藥物含量測定方法,應確定方法的定量限。常用的方法如下:
(1)直觀法
用已知濃度的被測物,試驗出能被可靠地定量測定的最低濃度和量。
(2)信噪比法
適用于能顯示基線噪音的分析方法,即將已知低濃度試樣測出的信號與空白樣品測出的信號進行比較,確定出能被可靠地定量的被測物質的最低濃度或量,一般以信噪比為10:1時相應濃度確定為定量限。
(3)基于響應值標準偏差和標準曲線斜率法
按照LOQ=10δ/S公式計算,式中LOQ為定量限、δ為響應值的偏差、S為標準曲線的斜率。δ可以通過下列方法測得: = 1 \* GB3 ①測定空白值的標準偏差; = 2 \* GB3 ②采用標準曲線的剩余標準偏差或是截距的標準偏差。以上計算方法獲得的定量數據須用含量相近的樣品進行驗證。附測定圖譜,說明試驗過程和定量結果,包括準確度和精密度驗證數據。
6. 線性
線性系指在設計的范圍內,線性試驗結果與試驗中被測物濃度直接呈比例關系的程度,是含量測定的基礎。線性試驗應至少包含5個不同濃度水平。以測得的響應信號與被測物濃度/含量作圖,觀察是否呈線性,再用最小二乘法進行線性回歸。必要時,響應信號可經數學轉換后進行線性回歸計算,或采用描述濃度/含量-響應關系的非線性模型。線性數據應至少列出回歸方程、相關系數、線性圖(或其他數學模型)。
7. 范圍
范圍系指分析方法能達到精密度、準確度和線性要求時的高低限濃度或量的區間。范圍根據分析方法的具體應用及其線性、準確度、精密度結果和要求確定。
選擇的方法應保證待測藥物濃度在其線性范圍內,必要時可對浸提液進行稀釋或濃縮。
8. 耐用性
耐用性系指在測定條件有小的變動時,測定結果不受影響的承受程度。如果測試條件要求苛刻,則應在方法中寫明,并注明可以接受變動的范圍。
9. 系統適用性
適用時,還應進行系統適用性考察。如高效液相色譜法的適用性試驗通常包括理論塔板數、分離度、靈敏度、拖尾因子、重復性等。
四、報告內容
研究報告一般包括試驗論述、試驗方案、試驗報告。其中,試驗論述應說明產品基本信息(材料組成、人體接觸途徑及接觸時間等)及藥物基本信息、試驗樣品選擇依據;浸提條件描述及其選擇依據(如適用);測定方法來源、適用性論述或測定方法驗證報告。試驗方案應包括浸提液制備(如適用)及測試方法、數據分析方案。試驗報告應報告詳細的測試方法(包括設備及試劑來源、樣品制備步驟,必要時附圖)、測試結果、數據處理、結論及典型性圖譜等。
五、參考文獻
[1]國家藥監局關于藥械組合產品注冊有關事宜的通(國家藥品監督管理局通告2021年第52號)[Z].2021.
[2]《冠狀動脈藥物洗脫支架臨床前研究指導原則》(國家藥品監督管理局通告2018年第21號)[Z].2018.
[3]《宮內節育器注冊技術審查指導原則》(國家藥品監督管理局通告2019年第25號)[Z].2019.
[4]GB/T 39381.1-2020《心血管植入物 血管藥械組合產品 第1部分:通用要求》[S].
[5]ISO/TR 12417-2:2017,Cardiovascular implants and extracorporeal systems - Vascular device-drug combination products - Part 2: Local regulatory information [S]
[6]ASTM F2394 - 07(2017) Standard Guide for Measuring Securement of Balloon Expandable Vascular Stent Mounted on Delivery System [S]
[7]《中國藥典》2020版 四部(國家藥典委員會)
[8]《醫療器械已知可瀝濾物測定方法驗證及確認注冊技術審查指導原則》(國家藥品監督管理局通告2019年第78號)[Z].2019.
[9]Guidelines for Single Laboratory Validation of Chemical Methods for Dietary Supplements and Botanicals(AOAC)
[10]Validation of Analytical Procedures:Text and Methodology Q2(ICH Harmonised Tripartite Guideline)
[11] GB/T 27417-2017《合格評定 化學分析方法確認和驗證指南》[S].
六、起草單位
國家藥品監督管理局醫療器械技術審評中心

%22/%3E%3C/g%3E%3C/g%3E%3C/svg%3E)


%22%3E%3Cg id=%22%E7%BC%96%E7%BB%84-13%22 transform=%22translate(30.000000, 2950.000000)%22%3E%3Cg id=%22%E7%BC%96%E7%BB%84-3%22 transform=%22translate(15.000000, 950.000000)%22%3E%3Cg id=%22%E7%BC%96%E7%BB%84-27%22 transform=%22translate(40.000000, 215.000000)%22%3E%3Cg id=%22%E7%BC%96%E7%BB%84-10%22 transform=%22translate(0.000000, 144.000000)%22%3E%3Crect id=%22%E7%9F%A9%E5%BD%A2%22 fill=%22%23d8d8d8%22 opacity=%22.01%22 x=%220%22 y=%220%22 width=%2232%22 height=%2232%22/%3E%3Cg id=%22ios-call%22 opacity=%22.4%22 transform=%22translate(4.800000, 4.800000)%22%3E%3Cmask id=%22mask-2%22 fill=%22%23fff%22%3E%3Cuse xlink:href=%22%23path-1%22/%3E%3C/mask%3E%3Cg id=%22Clip-2%22/%3E%3Cpath d=%22M21.739546 17.5430608C20.882919 16.6863974 18.8257249 15.3985249 17.8117151 14.9147941 16.6345314 14.3495128 16.2032859 14.3611681 15.3699694 14.9614517c-.693453.5012501-1.1421814.9673158-1.9406413.7926322C12.631014 15.5850459 11.057587 14.3903428 9.53067187 12.8692553c-1.52676943-1.5267694-2.71564488-3.10019644-2.8846465-3.89865628-.16903805-.80414178.29720975-1.2470426.7924865-1.9406413C8.03875902 6.1966413 8.05627837 5.76539577 7.48516943 4.58821203 7.0014387 3.56837463 5.7193574 1.51704455 4.85690276.660381138 4.00023935-.196245854 3.80789073-.00972487805 3.33596098.159276748c0 0-.69942635.279726829-1.39287935.740001301C1.08641821 1.47049626.608515122 1.94829008.270621138 2.66505366c-.3322120323.7168-.7168 2.0513665 1.241214962 5.53626016C3.09109073 11.0161015 4.64135285 13.1489821 6.94323512 15.4451824L6.95489041 15.4568377c2.30188227 2.3019187 4.42893529 3.8519987 7.24372289 5.4312898 3.4849301 1.9581242 4.8194602 1.573427 5.5362602 1.2413606C20.4516735 21.7972397 20.9295766 21.3193366 21.5006855 20.456882 21.9610693 19.7634289 22.2407961 19.0641119 22.2407961 19.0641119 22.4098341 18.5920364 22.6020371 18.3996878 21.739546 17.5430608z%22 id=%22Fill-1%22 fill=%22%23000%22 mask=%22url(%23mask-2)%22/%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
%22%3E%3Cg id=%22%E7%BC%96%E7%BB%84-13%22 transform=%22translate(30.000000, 2950.000000)%22%3E%3Cg id=%22%E7%BC%96%E7%BB%84-3%22 transform=%22translate(15.000000, 950.000000)%22%3E%3Cg id=%22%E7%BC%96%E7%BB%84-27%22 transform=%22translate(40.000000, 215.000000)%22%3E%3Cg id=%22%E7%BC%96%E7%BB%84-10%22 transform=%22translate(0.000000, 206.000000)%22%3E%3Crect id=%22%E7%9F%A9%E5%BD%A2%22 fill=%22%23d8d8d8%22 opacity=%22.01%22 x=%220%22 y=%220%22 width=%2232%22 height=%2232%22/%3E%3Cg id=%22ios-mail%22 opacity=%22.4%22 transform=%22translate(4.800000, 8.000000)%22 fill=%22%23000%22%3E%3Cpath d=%22M22.2035796 1.96720174 16.4132132 8.05344902C16.3701285 8.09790889 16.3701285 8.16447722 16.4132132 8.20893709l4.0521802 4.45365731c.274484099999999.2832104.274484099999999.738603.0 1.0216746C20.3308589 13.8230976 20.1478583 13.8953579 19.9703736 13.8953579 19.7927207 13.8953579 19.6097201 13.8230976 19.4751856 13.684269L15.4391495 9.24730586C15.3961994 9.20298482 15.3261742 9.20298482 15.2830559 9.24730586L14.298364 10.2802603c-.8233514.8607722-1.9158054 1.3382733-3.0889129 1.3438265C10.0201994 11.6296399 8.89004204 11.1132321 8.05592793 10.2413536L7.10877117 9.24730586C7.06568649 9.20298482 6.9957958 9.20298482 6.95271111 9.24730586L2.91667508 13.684269c-.13453454.1388286-.31743424.2110889-.49505346.2110889-.17761922.0-.36051892-.0722602999999999-.49505345-.2110889C1.65211772 13.4011974 1.65211772 12.9458048 1.92656817 12.6625944L5.97878198 8.20893709c.03756877-.044459869999999.03756877-.1110282.0-.155488069999999L.183000601 1.96720174C.112975375 1.89494143.0 1.94498915.0 2.04491106V14.2230629C0 15.2004165.774885285 16.0000347 1.72200841 16.0000347H20.6644372C21.611594 16.0000347 22.3866138 15.2004165 22.3866138 14.2230629V2.04491106C22.3866138 1.94498915 22.2680889 1.90052928 22.2035796 1.96720174z%22 id=%22Fill-1%22/%3E%3Cpath d=%22M11.1933069 10.1747852C11.9897177 10.1747852 12.7376961 9.85266811 13.2973598 9.264L21.7407135.395626898C21.4448721.15132321 21.0788372.00694143167 20.6752336.00694143167H1.71662703c-.40360361.0-.774885288.14438177833-1.065513516.38868546633L9.09446727 9.264C9.64878318 9.84711497 10.3968625 10.1747852 11.1933069 10.1747852z%22 id=%22Fill-2%22/%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/svg%3E)